Certains nocicepteurs sont qualifiés
de "silencieux" parce qu'ils ne répondent normalement
à aucun stimulus chimique, thermique ou tactile. Mais lors d'une blessure
avec inflammation, ils peuvent se "réveiller" et produire des
potentiels d'action suite à l'abaissement de leur seuil d'activation par
les différentes molécules inflammatoires. Plusieurs nocicepteurs
viscéraux sont des nocicepteurs silencieux.
Que ce soit pour la sensibilisation
centrale ou périphérique, on distingue au moins deux grands mécanismes
par lesquels la sensibilité du neurone va être augmentée
: un premier qui s'installe en quelques minutes, mais qui n'est que transitoire;
et un second qui apparaît plus lentement, en terme de jours, mais dure plus
longtemps. Ces mécanismes sont les mêmes qui ont été
décrits pour d'autres phénomènes de mémorisation au
niveau cellulaire, notamment la potentialisation
à long terme.
Dans le premier cas, il s'agit de changements
apportés à des protéines membranaires qui existent déjà.
Ces protéines chargées de la transduction du stimulus nociceptif
en influx nerveux se font par exemple phosphoryler, c'est-à-dire ajouter
un groupement phosphate par des enzymes comme les kinases. La phosphorylation
induit un changement de forme de la protéine qui deviendra par exemple
plus perméable à certains ions. Dans le cas du récepteur
canal AMPA du glutamate par exemple (impliqué dans la sensibilisation
centrale), la phosphorylation va augmenter sa probabilité et sa durée
d'ouverture, permettant ainsi plus d'ions sodium d'entrer dans le neurone, et
modifiant ainsi le potentiel de membrane dans le sens d'un abaissement global
du seuil d'excitabilité du nocicepteur.
Si le signal nociceptif
persiste, l'expression de gènes ou le rythme de traduction
de leur ARNm en protéine peut alors être augmenté. S'il
s'agit de nouveaux récepteurs, ces nouvelles protéines seront alors
acheminées jusqu'au bouton terminal de l'axone où elles contribueront
elles aussi à abaisser le seuil d'excitabilité et donc à
sensibiliser le neurone.
De nombreuses substances contribuent
à la sensibilisation des nocicepteurs. Le
facteur de croissance des nerfs (ou NGF), sécrété
par des fibroblastes (cellules de soutien dans le tissu conjonctif) et des cellules
de l'épiderme après leur stimulation par l'interleukine-1 sur le
site de l'inflammation, est l'un de ceux-là. Son récepteur spécifique
TrkA se retrouve environ chez 50% des nocicepteurs. L'activation de ce
récepteur conduit à la phosphorylation des résidus tyrosine
de sa partie intracellulaire et entraîne également la phosphorylation
intracellulaire d'autres molécules comme les récepteurs TRPV1 (voir
l'encadré à droite), un phénomène qui pourrait rendre
compte de l'hyperalgésie à la chaleur induite par le NGF.
Le
NGF pourrait aussi contribuer à une sensibilisation à plus long
terme par sa capacité à moduler l'expression de gènes comme
ceux de TRPV1 ou P2X3. On sait en effet que l'ensemble qu'il forme avec son récepteur
TrkA peut être internalisé par endocytose dans des vésicules
à l'intérieur de la fibre nerveuse. Puis, par transport rétrograde
dans l'axone, le complexe NGF/TrkA peut remonter jusqu'au noyau du nocicepteur
où il active la synthèse de nombreux peptides comme la substance
P et le CGRP. Il peut aussi promouvoir la synthèse de nouveaux récepteurs
pour les peptides algogènes sécrétés dans le foyer
inflammatoire comme les récepteurs à la bradykinine, les récepteurs
vanilloïdes VR1, etc.
LES MOLÉCULES QUI PRODUISENT
LA DOULEUR
La douleur est un
mécanisme essentiel
à notre survie. Elle ne survient normalement qu'en présence
de stimuli intenses qui sont potentiellement ou effectivement dommageables pour
l'organisme. Ceux-ci activent des fibres nerveuses à haut seuil, les nocicepteurs,
qui transmettent par des
voies ascendantes multiples le signal douloureux jusqu'au cerveau.
Mais
il arrive malheureusement que cette
douleur aiguë devienne chronique suite à une lésion tissulaire
entraînant de l'inflammation ou des dommages au système nerveux lui-même
(douleur
neuropathique). Les deux peuvent conduire à des douleurs surgissant
spontanément sans stimuli périphériques apparents ou encore
à une hypersensibilité aux stimuli périphériques.
L'hypersensibilité qui survient après une blessure n'est
pas mauvaise en soi. Elle est même adaptative dans la mesure où elle
aide la guérison en empêchant tout contact avec le tissue lésé.
Mais cette hypersensibilité persiste parfois au-delà de la guérison.
Dans ce cas, la douleur engendrée n'a plus aucun bénéfice
et est une manifestation de changements pathologiques dans le système nerveux.
Comprendre ce qui produit ces changements revêt donc une importance
cruciale pour soulager les douleurs chroniques. Deux mécanismes principaux
sont impliqués, soit la sensibilisation centrale et la sensibilisation
périphérique. Dans les deux cas, le terme général
de "sensibilisation" signifie une augmentation de l'excitabilité
des nocicepteurs, peu importe les mécanismes sous-jacents. Les nocicepteurs
réagissent donc à une surstimulation en devenant plus sensibles
contrairement à la plupart de nos autres récepteurs sensoriels qui
deviennent moins sensibles avec des stimulations répétées.
Dans la zone affectée, des
mécanorécepteurs sensibles à des stimuli tactiles légers
vont alors se mettre à activer des neurones de la moelle épinière
qui ne répondent normalement qu'à des stimuli nociceptifs. Le gain
du système se trouvant ainsi augmenté, le simple frôlement
d'un vêtement sur la peau peut produire une sensation douloureuse. Ce phénomène
appelé allodynie n'est pas la seule conséquence possible
d'une sensibilisation centrale. La plus grande sensibilité que l'on ressent
autour d'une blessure, dans une zone de tissus pourtant intacts, est aussi due
à une sensibilisation centrale et peut mener à l'établissement
de douleurs
chroniques.
Il se crée alors une espèce
de "mémoire de la douleur" (ou "wind up", en anglais)
qui peut être temporaire ou plus permanente selon que les modifications
moléculaires sous-jacentes sont de simples phosphorylations ou des changements
plus profonds au niveau de l'expression des gènes (voir encadré).
Ces modifications rendent aussi moins efficaces les
contrôles descendants utilisant les
opiacés pour réduire la douleur.
Une telle augmentation
de la réponse nociceptive à un stimulus donné n'est pas le
seul mécanisme pouvant mener à une hypersensibilité. L'abaissement
du seuil d'excitation des nocicepteurs eux-mêmes en est un autre. On parle
alors de sensibilisation périphérique pour décrire
ce qui arrive à ces fibres nerveuses qui deviennent plus sensibles qu'elles
ne l'étaient auparavant. Un exemple classique est le changement de sensibilité
qu'on expérimente après un coup de soleil. L'eau chaude mais habituellement
non douloureuse de la douche produira à l'endroit du coup de soleil une
sensation de brûlure.
Encore ici, divers mécanismes, certains
rapides, d'autres plus lents (voir encadré), vont contribuer à rendre
les terminaisons nerveuses des nocicepteurs de cette région plus sensibles.
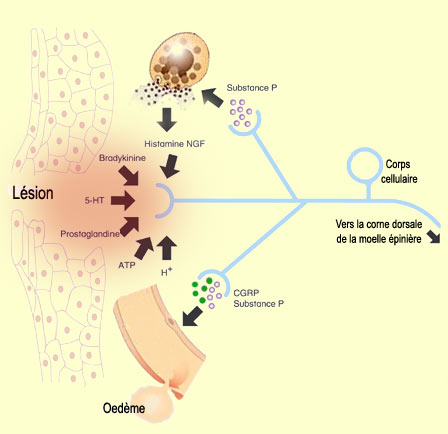
En effet, lorsque notre corps subit un assaut suffisant pour créer une
lésion, les cellules endommagées libèrent leur contenu dans
l'espace extracellulaire. Cette véritable "soupe de molécules"
va déclencher la sécrétion d'autres molécules dans
un processus connu sous le nom d'inflammation. En moins de 15 ou 30 secondes,
une rougeur et une chaleur dues à une vasodilatation des vaisseaux sanguins
apparaît autour de la blessure. Cette réponse inflammatoire, produisant
aussi oedème et libération de substances chimiques, atteint son
maximum 5 à 10 minutes plus tard.
Les molécules impliquées
dans ces réactions biochimiques locales proviennent de différentes
sources, mais l'origine première vient des cellules endommagées
elle-même. La lyse cellulaire libère par exemple la plupart du temps
des ions potassium en grande quantité. Et il y a une bonne corrélation
entre la concentration de potassium et le degré de douleur ressenti.
Même
chose pour la concentration extracellulaire d'ions H+, qui contribuent
eux-mêmes à activer directement des canaux ioniques au niveau de
certains nocicepteurs. Ce type de mécanisme sera par exemple responsable
des douleurs musculaires liées à la production d'ATP en condition
d'anaérobie qui génère de l'acide lactique lors d'exercices
particulièrement intenses.
De façon similaire, l'ATP
provenant des cellules lésées contribue à la dépolarisation
de certains nocicepteurs en activant directement des canaux ioniques dépendants
de l'ATP.
Après
une lésion tissulaire, les tissues environnants libèrent également
des substances telles que la bradykinine (l'un des plus puissants agents
algogènes connus), l'histamine et les prostaglandines. En
se fixant sur des récepteurs qui leur sont spécifiques sur la membrane
cellulaire des nocicepteurs (voir l'encadré ci-bas), ces molécules
déclenchent des potentiels d'action dans les fibres nociceptives.
L'aspirine
et les autres anti-inflammatoires non stéroïdiens sont le traitement
de référence de ces phénomènes d'hyperalgie,
du fait de leur action inhibitrice sur les enzymes impliquées dans la production
des prostaglandines.
Par un phénomène connu sous le nom de
"réflexe d'axone", les nocicepteurs vont aussi libérer
de la substance P dans leurs collatérales en périphérie,
c'est-à-dire vers les régions avoisinant le traumatisme. Cette libération
en périphérie, atypique puisqu'elle va dans le sens contraire de
normale pour un neurone sensoriel (efférent plutôt qu'afférent),
étend et amplifie la douleur en provenance de la zone lésée.
Cette libération de substance P va d'ailleurs amener certaines cellules
comme les mastocytes à libérer eux aussi de l'histamine, provoquant
ainsi une activation supplémentaire des fibres nociceptives dans cette
région élargie. De là l'efficacité des crèmes
qui bloquent les récepteurs de l'histamine (antihistaminiques) pour réduire
ces réactions inflammatoires douloureuses.
Outre la libération
périphérique de substance P par les nocicepteurs, ceux-ci libèrent
également le peptide relié au gène de la calcitonine
(calcitonin gene-related peptides ou CGRP, en anglais). Comme la substance P,
le CGRP présente une activité vasodilatatrice à la fois directement,
par son activité sur les cellules musculaires lisses, et indirectement
en favorisant la libération d'histamine par les mastocytes. Et cette dilatation
locale des capillaires produisant l'oedème favorisera à son tour
la libération de bradykinine.
À cela s'ajoutent bien d'autres
substances encore, comme la sérotonine libérée par
les plaquettes sanguines qui augmente elle aussi la perméabilité
des capillaires sanguins et contribue ainsi à la réaction inflammatoire.
Ou encore lefacteur
de croissance des nerfs(ou NGF), dont on connaît l'importance
pour le développement et la survie des neurones, et qui joue également
un rôle dans les processus inflammatoires (voir encadré).
On
voit donc comment ces mécanismes biochimiques sont complexes et comment
ils agissent en même temps sur deux fronts: en activant directement les
nocicepteurs, mais aussi en abaissant leur seuil d'activation, phénomène
à la base de la sensibilisation périphérique.
Les nocicepteurs possèdent de nombreux
récepteurs et canaux transmembranaires responsables de la transduction
des stimuli chimiques, mécaniques ou calorifiques. Ceux-ci, en modifiant
la conformation de leur molécule cible, vont altérer la conductance
membranaire et donc induire des courants locaux. La sommation de ces courants
locaux pourra ensuite déclencher des potentiels d'action si elle est suffisante.
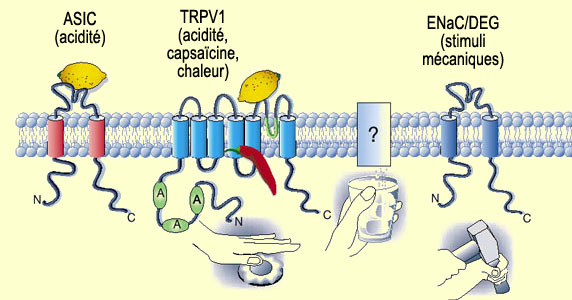
Les canaux TRP (de "Transient Receptor Potential", en
anglais) sont sensibles à des stimuli nociceptifs de différentes
natures. Ce sont en quelque sorte des "généralistes" qui
occupent le haut du pavé tant dans la nociception que dans d'autres détecteurs
sensoriels. Cette protéine
canal formée de six sous-unités transmembranaires laisse entrer
du calcium et du sodium dans le nocicepteur. Le sous-type TRPV1 (aussi appelé
récepteur vanilloïde, VR1) est par exemple sensible à la capsaïcine,
au pH bas créé par les protons extracellulaires (l'acidité)
et à la chaleur. Il s'agit donc d'un véritable intégrateur
de stimuli chimiques et physiques qui pourrait aussi être activé
par différentes protéines kinases. Celles-ci agiraient par l'entremise
de voies biochimiques distinctes dont les détails sont loin d'être
complètement élucidés.
À l'opposé, des
récepteurs comme les ASIC (pour "Acid-Sensing Ion Channels",
en anglais) sont des "spécialistes" qui ne répondent qu'à
un seul type de stimulus, en l'occurrence ici les protons extracellulaires. Ceux-ci
sont relâchés avec le contenu des cellules lésées ou
encore produits par la respiration anaérobique, par exemple sous forme
d'acide lactique qui rend les muscles endoloris. Un même type de stimulus
peut donc interagir avec de multiples récepteurs, comme le montre la capacité
des protons extracellulaires à activer non seulement les TRPV1, mais aussi
les ASIC.
La transformation des étirements et des déformations
mécaniques douloureuses des tissus est encore mal comprise. On pense que
des protéines de la famille ENaC/DEG agirait comme transducteur
mécanique non seulement dans les nocicepteurs
A delta, mais aussi dans les mécanorécepteurs. D'autres candidats
figurent aussi parmi les protéines TRP.
D'après
: Julius et Basbaum, Nature 2002.
De nombreux autres récepteurs
modulés par des substances émises par les réactions inflammatoires
vont contribuer à la génération de la douleur. Ainsi, l'ATP
extracellulaire produite par l'inflammation va se fixer aux récepteurs
purinergiques (par exemple P2X3). Ou encore l'activation des récepteurs
aux prostaglandines (PGE2), à la bradykinine (B1R and B2R)
et à la substance P (NK1), par leurs ligands respectifs, va aussi
contribuer à la réaction inflammatoire.
Cette activation
déclenchera souvent une cascade biochimique complexe comme dans le cas
du récepteur NK1 par exemple. Celui-ci est couplé à une protéine
G et induit l'activation de la phospholipase C qui va à son tour donner
de l'inositol tri-phosphate et du diacylglycérol en clivant son substrat,
le phosphatidylinositol di-phosphate ou PIP2.
La compréhension du
mode d'action de ces différents récepteurs intimement associés
à la nociception est essentielle pour ouvrir de nouvelles perspectives
thérapeutiques. En effet, les antidouleurs déjà existants
comme les
opiacés et les anti-inflammatoires non stéroïdiens (AINS)
agissent également sur des récepteurs en dehors des voies de la
douleur, produisant des effets secondaires indésirables.
La prise chronique d'opiacés
exogènes diminue les effets inhibiteurs sur les voies nociceptives
produits normalement par les ligands opioïdes endogènes. Cette diminution
s'explique entre autres par un découplage entre les récepteurs opioïdes
et les protéines G qui court-circuite la cascade de réactions biochimiques
subséquentes. Les récepteurs opioïdes eux-mêmes sont
aussi désensibilisés. Et la synthèse de leur ligand naturel,
les enképhalines par exemple, peut être diminuée. Enfin, la
morphologie même des neurones est modifiée par une diminution des
neurofilaments et l'inhibition du transport axonal.
Ces changements ne
sont pas sans altérer profondément l'activité nerveuse dans
les circuits de la douleur. Ils aboutissent à une réduction de l'efficacité
de notre système analgésique naturel aux endorphines ainsi qu'aux
phénomènes de tolérance
et de dépendance.
Un agoniste est une molécule
qui agit comme un ligand naturel en se liant au même récepteur que
lui. La morphine est par exemple un agoniste de la bêta-endorphine car elle
se fixe sur les récepteurs mu pour produire des effets similaires. La méthadone,
un composé synthétique utilisé pour amoindrir les syndromes
du sevrage
aux opiacés comme l'héroïne
ou la morphine, est également un agoniste des récepteurs mu.
L'antagoniste
d'une molécule se fixe lui aussi sur le même récepteur que
cette molécule, mais sans produire son effet. Comme une clé rentrée
dans la mauvaise serrure, il bloque le récepteur en occupant son site actif
et prévient par le fait même toute liaison avec le ligand naturel.
C'est ainsi qu'agit la naloxone, l'antagoniste le plus connu des opiacés..
Celle-ci est par exemple utilisée par voie intraveineuse après
une overdose d'héroïne qui a fait chuter la fréquence respiratoire
à deux ou trois respirations par minute. En compétitionnant rapidement
avec l'héroïne présente dans le sang du patient pour l'occupation
des récepteurs opioïdes responsable de la dépression respiratoire,
la naloxone peut ramener en quelques secondes la fréquence respiratoire
à quinze à vingt par minute.
Plus anecdotique, mais aussi
révélateur, lorsqu'on injecte des antagonistes aux opioïdes
à des gens capables de manger de très forts piments de type jalapeno,
le plaisir des mangeurs se transforme rapidement en douleur atroce, leurs endorphines
ne pouvant plus faire leur travail.
Il existe aussi ce qu'on appelle
des agonistes partiels. Ceux-ci occupent eux aussi les récepteurs
du ligand et produit le même effet, mais d'intensité moindre. Des
doses croissantes d'agonistes partiels sont donc accompagnées d'effets
croissants, mais à un certain point l'effet plafonne malgré les
doses toujours croissantes. À ces fortes doses, l'agoniste partiel se comporte
un peu comme un antagoniste s'il est mis en présence du ligand naturel:
il déplace progressivement ce dernier des sites actifs des récepteurs
et en réduit d'autant son effet.
La buprénorphine est un
agoniste partiel des récepteurs opioïdes de type mu utilisé
comme la méthadone pour le traitement substitutif de la dépendance
aux opiacés.
Notre système opioïde
interne semble jouer un rôle dans
la dépendance psychique aux drogues. On a par exemple démontré
que des souris dépourvues de récepteurs opiacés de type "
mu " ne s'auto-administrent pasd'alcool
ou de cocaïne
alors que les souris normales utilisent abondamment ce dispositif permettant l'autostimulation.
Dans une autre expérience, des rats reçoivent une injection
de morphine
et sont ensuite placés dans un compartiment au mur coloré. Le jour
suivant, les rats reçoivent un placebo
et sont placés dans un autre compartiment au mur d'une couleur différente.
On continue ainsi plusieurs fois cette alternance et l'animal apprend ainsi à
associer un environnement spécifique avec les effets positifs de la morphine.
Après cette période de conditionnement, les rats sont replacés
(sans injection de drogue ni de placebo) dans l'un, puis dans l'autre des compartiments.
Lorsqu'un rat est placé dans l'environnement associé à l'injection
de morphine, on constate une augmentation de la concentration synaptique des enképhalines
dans le noyau
accumbens de son cerveau. Mais quand on le met dans le compartiment associé
à l'injection du placebo, c'est plutôt une diminution des enképhalines
que l'on observe.
Cette production d'enképhalines dans un
site clé du circuit de la récompense semble donc être
impliquée, du moins en partie, à l'anticipation d'une récompense.
Par ailleurs, rappelons que la dopamine,
neurotransmetteur grandement associé au plaisir, est libérée
sous l'influence de deux types de neuropeptides : les cholécystokinines
et… les enképhalines qui se fixent sur des récepteurs opioïdes
de type " mu " et " delta " !
LES
MOLÉCULES QUI DIMINUENT LA DOULEUR
Les substances opioïdes extraites de plantes
sont utilisées depuis des siècles pour soulager la douleur. Au début
des années 1970,
la découverte de récepteurs membranaires spécifiques
à ces molécules et, quelques années plus tard, des
peptides endogènes qui s'y lient , a jeté les bases de notre
compréhension des mécanismes complexes de notre système à
endorphines.
Les récepteurs aux endorphines constituent les éléments
clés qui permettent de comprendre l'action anti douleur des endorphines
ou des médicaments analgésiques opioïdes (voir la capsule outil).
Comme la plupart des récepteurs, il s'agit de grosses protéines
insérées dans la membrane cellulaire du neurone.
On distingue
quatre grandes familles de récepteurs opioïdes qui toutes sont formées
de protéines ayant 7 domaines transmembranaires. La partie exposée
au milieu extracellulaire possède un site spécifique dont la forme
est complémentaire à celle des substances opioïdes qui donc
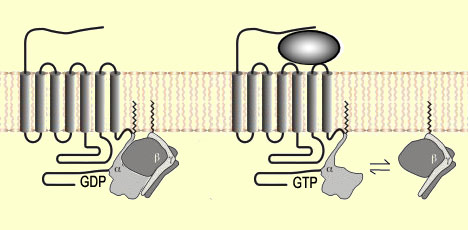
peuvent s'y fixer. Cette fixation provoque une modification de la forme du récepteur
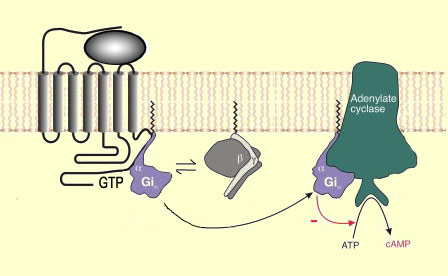
qui va activer, du côté intracellulaire, une protéine G formée
de trois sous-unités (alpha, bêta et gamma).
Cette
activation conduit au remplacement, sur la sous-unité alpha, de la molécule
de guanine diphosphate (ou GDP) qui y était liée par une molécule
de guanine triphosphate (ou GTP). La GTP induit à son tour la dissociation
de l'ensemble alpha, bêta et gamma en une sous-unité alpha et une
sous-unité bêta-gamma.
Chacune de ces deux entités contribuera
ensuite à la transduction du signal, c'est-à-dire le déclenchement
d'une cascade de réactions biochimiques (on parle aussi de " seconds
messagers ") à l'intérieur de la cellule suite à un
événement déclencheur à l'extérieur de la cellule.
Dans le cas de la fixation d'un peptide opioïde endogène ou d'une
substance opiacée d'origine externe sur un récepteur opioïde,
les effets sont généralement inhibiteurs sur l'activité nerveuse
de la cellule.
Les mécanismes
les mieux connus de ces inhibitions impliquent l'adénylate cyclase, une
enzyme qui transforme de l'ATP
en AMP cyclique. La diminution d'AMP cyclique, un second messager important qui
interagit avec plusieurs autres protéines, est à l'origine par exemple
de l'hyperpolarisation
neuronale observée suite à la fixation d'agoniste mu ou delta sur
les récepteurs opioïdes. Des canaux potassiques sont ici affectés
par l'AMP cyclique pour produire cette baisse d'excitabilité neuronale
qu'on appelle hyperpolarisation.
Un autre effet produit par
la cascade de seconds messagers initiée par la protéine G concerne
les canaux calciques sensibles au voltage situés sur le bouton terminal
de l'axone (près de la synapse). L'effet ici est une diminution de perméabilité
au calcium de ces canaux qui entraîne une baisse de la libération
de neurotransmetteurs, notamment de la substance
P et du glutamate, présents dans les afférences sensorielles
nociceptives des fibres
C.
Cet affaiblissement de transmission de l'influx nerveux au niveau
présynaptique, tout comme l'élévation du seuil de déclenchement
des potentiels d'action dans le neurone post-synaptique (hyperpolarisation), va
diminuer l'activité globale dans les voies ascendantes de la douleur (voir
l'encadré) et par conséquent sa perception.
Dans chaque grande
famille de récepteurs opioïdes, on compte aussi différents
sous-types dont l'activation peut être sélective pour certains ligands.
Ceux-ci produiront des effets différents selon la nature particulière
du sous-type de récepteur auquel il se fixe, mais aussi selon le type de
neurone où l'on retrouve ces récepteurs.
On connaît
au moins quatre grandes familles de récepteurs opioïdes, désignés
respectivement par les lettres grecques " mu ", " delta ",
" kappa " et la quatrième par le sigle ORL-1 (pour " Opioid
Receptor-Like ", en anglais).
Les
récepteurs de type delta furent les premiers à être décrits
vers le milieu des années 1970. Ce sont les
enképhalines qui ont la plus grande affinité avec les récepteurs
delta et elles sont pour cette raison considérées comme leur ligand
naturel. Les enképhalines peuvent cependant aussi se fixer aux récepteurs
mu et kappa, mais avec une affinité moindre.
Difficiles à caractériser au début
à cause de la faible durée de vie des enképhalines, on connaît
un peu mieux les récepteurs delta depuis que des peptides qui leur sont
spécifiques ont pu être synthétisés. On sait par exemple
que l'activation des récepteurs delta produit une analgésie, quoique
moindre que celle des récepteurs mu. Mais celle-ci semble cependant s'accompagner
aussi de moins d'effets secondaires indésirables, notamment au niveau de
la dépression respiratoire, de la constipation et de la tolérance.
D'où l'intérêt dans la recherche sur la douleur
chronique pour les agonistes (voir l'encadré) sélectifs du récepteur
delta.
Une autre caractéristique intéressante du récepteur
delta est son effet régulateur sur l'humeur. Des souris mutantes ne possédant
pas le gène du récepteur delta ont démontré un degré
d'anxiété plus élevé et des comportements dépressifs.
Encore ici, des agonistes spécifiques pourraient s'avérer précieux
dans le traitement des troubles de l'humeur.
Les récepteurs delta
sont bloqués par la naloxone, mais celle-ci s'y fixe avec une moins grande
affinité que sur les récepteurs mu.
Les récepteurs
de type mu ont comme ligand naturel la bêta-endorphine,
second peptide opioïde identifié après les enképhalines.
Celles-ci s'y fixent d'ailleurs aussi avec une bonne affinité, ce qui n'est
pas le cas des dynorphines dont l'affinité pour les récepteurs mu
est faible.
C'est également sur
les récepteurs de type mu que se fixent préférentiellement
la morphine et les autres dérivés opioïdes exogènes.
Ces analgésiques morphiniques produisent cependant des effets secondaires
indésirables (dépression respiratoire, constipation, tolérance,
etc) principalement par l'entremise du récepteur mu.
Les
techniques de la biologie moléculaire ont permis d'identifier de multiples
variantes du gène du récepteur mu. Un premier sous type, le récepteur
mu-1, est davantage associé à l'effet analgésique et un autre,
le récepteur mu-2, aux effets respiratoires et de motilité intestinale.
On a également proposé un rôle des récepteurs
mu dans l'attachement mère enfant et dans le circuit
de la récompense. Cette dernière implication pourrait d'ailleurs
être à l'origine des comportements de dépendance
induits par des substances comme l'éthanol, la nicotine, l'héroïne
ou la morphine. Cette dépendance ne s'installe d'ailleurs pas chez les
souris dont les récepteurs mu ont été désactivés.
La distribution des récepteurs mu dans l'organisme correspond aux
effets qu'ils produisent. Ceux-ci sont par exemple largement présents dans
la région des centres respiratoires du tronc cérébral. Ou
encore au niveau pré-synaptique dans la substance
grise périaqueducale et la corne dorsale de la moelle épinière
où ils contribuent à l'inhibition
descendante de la douleur.
Ce contrôle descendant, à l'œuvre
aussi dans l'effet
placebo, est d'ailleurs bloqué par la naloxone, un antagoniste puissant
des récepteurs de type mu.
Les
récepteurs de type kappa ont la plus grande affinité pour la
dynorphine,
peptide opioïde découvert à la fin des années 1970.
Encore ici, l'affinité des différents sous-types de dynorphine pour
les récepteurs kappa varie cependant. Comme les autres récepteurs
opioïdes, les récepteurs kappa induisent une analgésie, mais
provoquent également des nausées, de la dysphorie et autres effets
psychiques indésirables, ce qui ajoute aux difficultés liées
au développement d'agoniste de synthèse pour ce type de récepteur.
On trouve aussi de nombreux liens entre le
stress chronique, ses effets néfastes sur la santé et les dynorphines.
Le système à dynorphine, et donc en bout de ligne ses récepteurs
kappa, contrôlerait certains circuits neuronaux reliés au stress,
produisant ainsi les effets dysphoriques qui accompagnent cet état de tension
permanente.
De façon plus générale, de plus en plus
de données montrent que l'activation des récepteurs kappa produit
des effets qui s'opposent à ceux qui découlent de l'activation des
récepteurs mu (analgésie, tolérance, récompense, mémoire,
etc.). Ces effets opposés impliqueraient aussi une localisation des deux
types de récepteur sur différentes catégories de neurones
dans ces circuits utilisant les peptides opioïdes.
Le plus récent
récepteur opioïde à avoir été mis en évidence
est le récepteur à la nociceptine ou ORL-1 (pour " opioïd
receptor-like ", en anglais). Ce récepteur a une grande affinité
pour la nociceptine (aussi appelée orphanine)
tout en ayant très peu pour les trois autres grandes familles d'endorphine.
Et inversement, les peptides opioïdes autres que la nociceptine ne se fixent
guère sur les récepteurs ORL-1.
Voilà
qui est assez singulier pour un récepteur qui montre une grande homologie
de structure avec les trois autres classes de récepteurs opioïdes.
Cette singularité se manifeste aussi au niveau des fonctions du récepteur
ORL-1 qui, selon la dose et le site d'action, peut être tantôt un
antagoniste aux effets des opiacés, tantôt un analgésique.
De plus, l'activation du récepteur ORL-1 affecterait directement ou indirectement
(par l'entremise du GABA), le taux de dopamine.
La pharmacologie du récepteur ORL-1 semble donc très complexe selon
les données disponibles actuellement.
Les endorphines et leurs récepteurs
sont très largement distribués dans le système nerveux, tant
au niveau supraspinal, spinal que périphérique. Ils sont particulièrement
représentés dans les régions impliquées dans le
contrôle descendant de la douleur.
Au niveau supraspinal,
la présence de récepteurs opioïde est très documentée
dans la substance
grise périaqueducale: les études d'autoradiographie ont montré
leur présence dans la substance grise périaqueducale; la micro-injection
de morphine dans cette structure s'est avérée profondément
analgésique, de même que sa stimulation électrique; cet effet
pouvait être bloqué par la naloxone, un antagoniste des récepteurs
opioïdes (voir le deuxième encadré à gauche), etc.
Ce
sont les
stimuli nociceptifs en provenance de la moelle épinière ainsi
que les connexions de nombreuses autres structures du tronc cérébral
et des centres supérieurs qui peuvent normalement déclencher la
libération d'endorphine dans la substance grise périaqueducale.
D'autres
sites d'action supraspinaux probables pour leur libération incluent la
formation réticulée, la substance noire, les noyaux du raphé,
l'hypothalamus, l'hippocampe, le noyau caudé, l'amygdale et le cortex préfrontal
ventral, etc.
Au niveau spinal, les récepteurs opioïdes
sont situés au niveau des terminaisons axonales des fibres C et sur les
corps cellulaires des neurones nociceptifs des couches superficielles de la corne
dorsale de la moelle épinière.
Plusieurs interneurones
localisés à proximité des terminaisons axonales des fibres
C ou A delta dans ces couches superficielles de la moelle épinière
émettent des enképhalines comme neurotransmetteur. Celles-ci peuvent
être relâchées par l'activation des fibres sérotoninergiques
en provenance de la formation réticulée. La
porte d'entrée du signal nociceptif peut donc être fermée
soit par la diminution de substance P ou de glutamate émis par les fibres
C ou A delta, soit par la baisse d'excitabilité des neurones nociceptifs
de la moelle épinière. Mais l'un comme l'autre résulte de
la fixation des enképhalines sur leurs récepteurs spécifiques.
Au niveau périphérique, des récepteurs
opioïdes ont été identifiés sur plusieurs terminaisons
de fibres sensorielles, notamment les fibres nociceptives de type C. Les trois
principaux types de récepteurs opioïdes sont produits dans les corps
cellulaires de ces neurones (situés dans les ganglions spinaux) et acheminés
par transport axonal jusqu'aux terminaisons périphériques.
L'effet
analgésique médié par ces récepteurs opioïdes
périphériques serait particulièrement important dans les
fibres nociceptives déjà
sensibilisées par une inflammation. Des lésions tissulaires
stimulent d'ailleurs l'expression des récepteurs opioïdes.
L'administration
d'un médicament opioïde par voie orale ou intraveineuse exercera donc
ses effets analgésiques à plusieurs niveaux. Le fait que des cellules
immunitaires expriment également des récepteurs opioïdes, indique
que ces effets pourraient être encore plus larges.