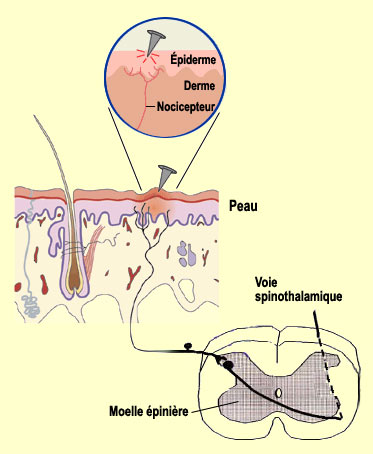
Les terminaisons libres des nocicepteurs sont
très nombreuses à la surface de notre corps, ce qui explique la
grande sensibilité de notre peau à la douleur. Ces nocicepteurs
de la peau sont situées tant dans l'épiderme (couche superficielle)
que dans le derme (couche profonde). Les nocicepteurs de la peau sont concentrés
dans les régions du corps les plus exposées aux blessures, comme
les doigts et les orteils. C'est la raison pour laquelle une écharde
ou éclat de verre sera plus douloureux lorsque logé dans un doigt
que dans la poitrine ou l'épaule.
Protégés par la
peau, les muscles comportent moins de terminaisons nerveuses qui sont réparties
de façon assez espacée et discontinue. Cela explique le caractère
diffus et mal localisable de la douleur musculaire (crampes, courbatures, etc.).
La
paroi des vaisseaux sanguins est pour sa part richement innervée,
les terminaisons libres sensibles aux stimuli nociceptifs étant situées
dans la couche interne des vaisseaux artériels et veineux.
Au niveau
osseux, la plupart des terminaisons libres se situent dans la moelle et
le périoste où elles forment un réseau régulier. Tout
ce qui lèse l'organisation de l'os, particulièrement de façon
brutale comme une fracture, causera donc de la douleur.
Aux articulations,
les récepteurs nociceptifs sont répartis surtout dans la capsule,
la synoviale, les ligaments et les tendons. Ils peuvent être activés
mécaniquement (étirements, déchirement, etc.) ou biochimiques,
comme lors d'un processus inflammatoire comme l'arthrite.
Les viscères,
protégés par la peau, les muscles et les os, comportent encore moins
de terminaisons nerveuses que les muscles. Les terminaisons libres y sont réparties
de façon à la fois lâche et très inégale. D'où
le caractère souvent vague et difficilement localisable de ce type de douleur,
qu'il s'agisse de l'inconfort d'une indigestion aux douleurs terribles d'une colique
néphrétique (ou rénale).
Les viscères les plus
innervés par les nocicepteurs sont surtout ceux qui sont creux (intestin,
vessie, utérus, etc.). Comme ils sont le prolongement interne de notre
environnement externe, ils sont davantage susceptibles d'être en contact
avec des agents potentiellement dangereux et nécessitent donc une surveillance
adéquate. À l'inverse, les viscères pleins (poumons, foie,
rate) possèdent moins de terminaisons libres et sont peu sensibles à
la douleur. Par conséquent, ils peuvent se détériorer sans
qu'on en ait trop conscience.
LES MOLÉCULES QUI PRODUISENT LA DOULEUR
La sensation du toucher
prend naissance dans des récepteurs spécialisés situés
dans notre peau. Ces mécanorécepteurs sensibles à
des pressions et des étirements faibles ou modérés envoient
des messages nerveux que notre système
nerveux central interprète comme des sensations tactiles.
Mais
lorsque ces pressions mécaniques deviennent fortes au point de menacer
l'intégrité de nos tissus ou lorsque ceux-ci sont carrément
endommagés, ce sont les récepteurs à la douleur, ou nocicepteurs,
qui prennent la relève.
Il s'agit de terminaisons nerveuses
libres c'est-à-dire les extrémités d'axones
dépourvues de myéline.
Ces terminaisons axonales très arborisées sont celles des fibres
de type A delta et C. Elles se retrouvent aussi bien dans les tissus cutanés,
musculaires, vasculaires, articulaires, osseux ou viscéraux (voir l'encadré).
Bref pratiquement partout sauf
à l'intérieur même du cerveau ! La notion de nocicepteur
fait donc d'abord référence à une fonction (celle de ressentir
la douleur) plutôt qu'à des récepteurs spécialisés
(comme dans le cas des mécanorécepteurs).
Les nocicepteurs peuvent être activés
par toutes sortes de stimuli qui peuvent potentiellement altérer les tissus,
et pas seulement des stimulations mécaniques comme les pinçures,
les piqûres ou les morsures. Des températures extrêmes (voir
l'encadré), des chocs électriques, des conditions d'hypoxie (manque
d'oxygène) ou encore des expositions à des substances toxiques peuvent
également les activer. Si certains nocicepteurs sont plus sensibles à
un type de stimulus qu'à un autre, la plupart sont toutefois polymodaux,
c'est-à-dire qu'ils peuvent répondre à plus d'un type de
stimulus.
Quelle que soit sa nature, le stimulus doit
aussi atteindre une certaine intensité pour pouvoir activer les nocicepteurs
(voir l'encadré). Le seuil d'activation de ces derniers est donc plus élevé
que celui des mécanorécepteurs. Les nocicepteurs peuvent donc coder
l'intensité des stimulations douloureuses en modulant leur réponse
selon l'intensité du stimulus. Et au niveau cutané, le seuil de
leur activation correspond à celui de la sensation douloureuse perçue
par le sujet.
C'est grâce à différents
types de canaux ioniques situés à travers leur membrane cellulaire
que les nocicepteurs sont sensibles à cette vaste gamme de stimulations
douloureuses. Ces stimulations peuvent être directes, comme dans le cas
des fortes pressions mécaniques qui déforment les membranes et déclenchent
des influx nerveux. C'est par exemple la punaise qui nous pique le pied, mais
sans percer la peau.
Mais si la punaise perce la peau
et endommage les tissus, les cellules altérées vont libérer
localement certaines substances chimiques qui vont, de manière indirecte,
stimuler aussi les nocicepteurs. Ces
molécules dites algogènes libérées par les tissus
endommagés ou enflammés, peuvent être des enzymes comme
la bradykinine, des neurotransmetteurs comme la sérotonine,
ou encore des hormones comme la prostaglandine. Le message douloureux peut également
naître de la lésion d'une fibre nerveuse.
En
cas de stimulations fortes ou répétées, les nocicepteurs
sont le siège de phénomènes de sensibilisation
qui vont abaisser le seuil de réponse, augmentant le nombre d'influx nerveux
et la sensation douloureuse ainsi produite.
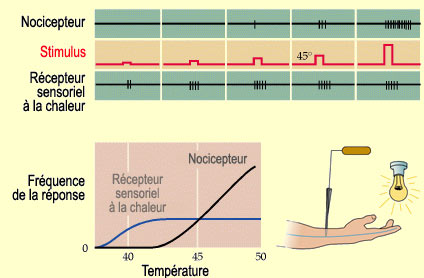
Une sensation de chaleur douloureuse n'est pas
produite par une activité nerveuse excessive des récepteurs actifs
lors d'une chaleur confortable. On a clairement démontré l'existence
de nocicepteurs thermiques distincts des récepteurs sensoriels à
la chaleur. Quand on augmente progressivement la température du stimulus,
on note une augmentation proportionnelle dans les récepteurs sensoriels
à la chaleur jusqu'à environ 45 degrés Celsius où
leur activité plafonne. À l'opposé, dans les nocicepteurs
thermiques, l'activité ne commence qu'après 40 degrés Celsius,
mais augmente ensuite proportionnellement à la chaleur, sans effet de plafonnement.
Or
c'est justement autour de 45 degrés Celsius les protéines commencent
à se dénaturer et nos tissus à être endommagés.
Et c'est à cette température que les gens décrivent une chaleur
comme devenant douloureuse, différents
facteurs pouvant bien entendu faire varier le seuil douloureux
de quelques degrés.
Les termes "opiacé" et "opioïde"
n'ont pas tout à fait la même signification.
On parle de substance
ou de médicament opiacé quand celui-ci contient de l'opium
ou ses dérivés comme la morphine ou la codéine. Il s'agit
de molécules qui ne sont pas des peptides (plutôt des alcaloïdes)
et qui proviennent d'une source extérieure au corps humain (les graines
de pavot ou des composés synthétiques).
Le terme opioïde
caractérise quant à lui un groupe de peptides endogènes exerçant
un effet physiologique semblable à celui de la morphine. Son usage tend
toutefois à se répandre pour designer toute substance (naturelle
ou synthétique, de nature peptidique ou non) qui agit sur les récepteurs
désignés justement comme… opioïdes !
Peu de temps après la découverte
de substances opioïdes naturelles dans le cerveau, celles-ci ont été
associées au phénomène de l'euphorie du coureur ("runner's
high" en anglais). Il s'agit d'une sensation intense de bien-être que
connaissent bien les coureurs de longue distance. Mais cette euphorie peut aussi
être ressentie par toute personne qui pratique une activité physique
d'intensité modérée pendant 20 ou 30 minutes.
Les
scientifiques étaient toutefois restés sceptiques quant au rôle
de morphines endogènes dans l'euphorie du coureur, entre autres parce qu'on
ne parvenait pas à empêcher son émergence avec des bloqueurs
aux récepteurs opioïdes comme la naloxone.
L'implication d'une
autre famille de molécules, les endocannabinoïdes, qui sont notre
"cannabis
endogène", a même été avancée pour expliquer
cette modification positive de l'humeur. Les endocannabinoïdes,
comme les endorphines, ont leurs propres récepteurs spécifiques
sur lesquels ils se fixent, tout comme le THC (la substance active du cannabis)
pour produire leurs effets. Ceux-ci contribuent à la modulation de la douleur.
Et comme l'humeur euphorique fait partie de ces effets, un lien avait été
établi avec l'euphorie du coureur après avoir constaté une
hausse de la concentration des endocannabinoïdes dans le corps après
un exercice soutenu.
Mais une étude publiée par des chercheurs
allemands en 2008 a remis les peptides opioïdes endogènes au centre
de ce phénomène. À l'aide d'un test psychologique, les scientifiques
ont d'abord évalué l'humeur de 10 personnes. Puis ils leur ont injecté
une substance radioactive permettant de révéler la présence
d'endorphines dans leur cerveau grâce à une technique d'imagerie
cérébrale, la tomographie par émission de positons, ou PET
scan.
Les 10 sujets, qui étaient des coureurs aguerris, ont ensuite
été courir pendant deux heures. Dès leur retour, on a réévalué
leur humeur et la distribution de la radioactivité, et donc la présence
d'endorphines, dans leur cerveau. Et ce qu'ont constaté les chercheurs,
c'est que plus le sentiment d'euphorie est intense, plus il y avait d'endorphines
dans une région de leur cerveau associée aux émotions, le
système
limbique et le cortex
préfrontal.
L'étude ne précise toutefois
pas le
type précis d'endorphine impliqué dans l'euphorie du
coureur, une clarification qui s'avérera nécessaire considérant
leur multitude et leurs différents effets. Mais un lien important semble
maintenant établi: les mêmes substances qui ont la capacité
d'atténuer
notre perception d'une douleur intense sont aussi en jeu
pour compenser (et même inverser !) l'inconfort physique dû à
un effort soutenu.
LES MOLÉCULES
QUI DIMINUENT LA DOULEUR
On utilise depuis des siècles l'opium et
différentes substances qui en sont dérivées, comme la morphine,
pour apaiser la douleur. Les tablettes cunéiformes de Sumer ou les idéogrammes
chinois louaient déjà les vertus de l'opium. Mais il a fallu attendre
les années 1970 pour se rendre compte que le cerveau humain produisait
lui aussi ses propres morphines endogènes. L'histoire des multiples découvertes
qui ont mené à cette conclusion est fascinante à plusieurs
égards.
D'abord au niveau de la démarche scientifique, où
des déductions justes ont permis d'amasser rapidement des indices. Puis
au niveau technique, où les chercheurs ont dû faire preuve de beaucoup
d'ingéniosité et de détermination étant donné
la faible concentration des molécules à isoler. Et enfin au niveau
de la sociologie du milieu scientifique, puisqu'elle a impliqué une véritable
course entre différents laboratoires, mais aussi une collaboration nécessaire
entre ceux-ci.
On savait donc au départ qu'une substance végétale,
l'opium (ou ses dérivés), avait un effet dans le corps d'un animal.
Or pour qu'une molécule agisse sur l'activité d'une cellule, la
règle générale est qu'elle doit se fixer sur des récepteurs
situés la plupart du temps sur la membrane de cette cellule. Et c'est cette
fixation, comme
une clé qui débarre une serrure, qui ouvre la porte à
une cascade de phénomènes biochimiques qui vont avoir un effet physiologique,
dans ce cas-ci un effet analgésique.
D'où la première
déduction que si les substances opiacées végétales
agissaient sur les cellules nerveuses animales, il fallait que celles-ci aient
des récepteurs aux opiacés comme on en avait identifié pour
d'autres
neurotransmetteurs. Ce raisonnement marqua le début de la saison de
la " chasse aux récepteurs " aux opiacés qui débuta
au début des années 1970, à une époque où la
neurochimie, la discipline qui s'intéresse à ce genre de chose,
commençait à peine à émerger comme discipline scientifique.
Convaincu que les
effets très ciblés de la morphine (contraction de la pupille,
diminution du rythme cardiaque, diminution de la douleur ressentie, etc) ne pouvaient
venir que de récepteurs qui lui étaient spécifiques, l'américain
Solomon Snyder commença ses recherches avec l'approche la plus directe.
Celle-ci consistait à mettre en contact de la morphine radioactive avec
des neurones et de voir, après lavage, si l'on pouvait détecter
des molécules radioactives attachées aux neurones. Si oui, c'était
le signe de la présence de récepteurs.
Malheureusement,
rien ne restait accroché aux neurones, même lorsque l'expérience
fut répétée avec de l'héroïne
au lieu de la morphine. Alors que la communauté scientifique commençait
à avoir des doutes sur l'existence de récepteurs aux opiacés,
Snyder, aidé de son étudiante graduée Candace Pert,
tenta une dernière fois sa chance, mais cette fois-ci avec une substance
connue pour bloquer les effets des opiacées, la naloxone.
Et
là, victoire, la naloxone radioactive restait bien fixée aux neurones
! Cela confirmait l'hypothèse déjà émise que la naloxone
bloquerait les effets de la morphine en se fixant sur des récepteurs, et
probablement ceux de la morphine eux-mêmes. Avec ce résultat et un
peu de recul, on comprit que contrairement à la naloxone qui reste littéralement
prise dans le récepteur pour le bloquer, la morphine ou l'héroïne
agissent en se fixant pour une très courte durée sur le récepteur.
Ces molécules ne pouvaient donc pas tenir assez longtemps sur les récepteurs
pour être détectées.
D'autres
chercheurs comme Eric Simon et Lars Terenius publièrent également
en 1973 des observations semblables confirmant la présence de récepteurs
aux opiacés dans le système nerveux central, ce qui généra
une grande excitation dans le domaine.
En effet, la présence
de récepteur aux opiacés voulait presque automatiquement dire qu'il
devait y avoir aussi une substance opiacée produite naturellement par le
cerveau pour s'y fixer. On imagine mal que que l'évolution ait mis en place
des serrures aussi spécifiques sans qu'il n'y ait des clés originales
pour les ouvrir (et pas seulement des copies fortuites venant du monde végétal).
Cette
idée que la morphine pourrait simplement mimer les effets d'une substance
déjà présente dans l'organisme était déjà
derrière la tête de plusieurs des scientifiques qui avaient travaillé
à isoler le récepteur. Mais elle avait été particulièrement
bien défendue par le biologiste d'origine allemande Hans Kosterlitz.
Celui-ci, travaillant à
l'université d'Aberdeen en Écosse, incita le sous-directeur de son
laboratoire, John Hughes, à démontrer l'existence d'une telle
morphine endogène. Facile à dire, mais plutôt compliqué
à faire avec les outils de l'époque. Hughes choisit donc un "
détecteur " à morphine endogène qui lui était
accessible : le canal déférent de la souris dont les contractions
étaient inhibées par la morphine qui s'y fixait sur des récepteurs
spécifiques. L'idée était donc d'appliquer des extraits de
cerveau sur cette préparation et de voir si les contractions cessaient.
Le
problème, c'est que certaines substances peuvent agir à des concentrations
aussi faibles que ce qui correspond à un gramme de substance dans dix millions
de litres d'eau. Les chances de succès apparaissaient donc infimes et John
Hughes dut investir les abattoirs d'Aberdeen pour y prélever des milliers
de cerveaux de porc pour son laboratoire. Certaines structures cérébrales
y étaient alors broyées, concentrées et appliquées
sur le canal déférent de souris.
Des indices de la présence
d'une substance endogène capable de se lier aux récepteurs opiacés
ont été obtenus dès 1973 par Hughes et le labo de Lars Terenius.
Puis, en mai 1974, ces résultats préliminaires ont été
communiqués et en décembre 1975, Hughes et Kosterlitz publiaient
la structure de deux substances qu'ils nommèrent enképhalines,
du grec " dans la tête ". Les deux substances en question étaient
des peptides,
c'est-à-dire de petites protéines
formées de quelques acides aminés.
Les deux enképhalines étaient formées de cinq acides aminés
et seul le dernier de la chaîne différait : c'était de la
méthionine pour la met-enképhaline et de la leucine pour la leu-enképhaline.
En
1976, les équipes de Choh Hao Li et de Roger Guillemin isolent
des peptides plus longs, les endorphines,
capables eux aussi de se lier aux récepteurs opiacés. De nombreux
autres peptides opioïdes endogènes comme les dynorphines,
furent ensuite isolés, de sorte qu'en 1992, on en comptait déjà
une vingtaine.
In 1971, John C. Liebeskind et ses collègues
publièrent d'étranges observations. La stimulation d'une région
du mésencéphale
appelé substance
grise périaqueduquale produisait une douleur chez l'animal,
mais l'arrêt de la stimulation où la diminution de son intensité
avait un effet analgésique. Cet article suggérait en outre que cet
effet était analogue à celui des médicaments opiacés.
Dès l'année suivante, ils confirmaient d'ailleurs leur intuition
en montrant que cette analgésie produite par stimulation électrique
pouvait être empêchée par une substance reconnue pour bloquer
les effets des opiacés, la naloxone.
Liebeskind
établit de nombreux autres parallèles entre le type d'analgésie
produite par ces stimulations et l'effet des médicaments opiacés,
pavant ainsi la voie à l'identification des mécanismes
de contrôle descendant de la douleur, des récepteurs
opioïdes et des premières morphines endogènes qui
allait se faire dans les années suivantes.