|
|
| Le financement de ce site est assuré par vos dons, merci! | |
|
|
|
|
|
|||||
|
|
|||||||
|
|
|
|


|
|
| |
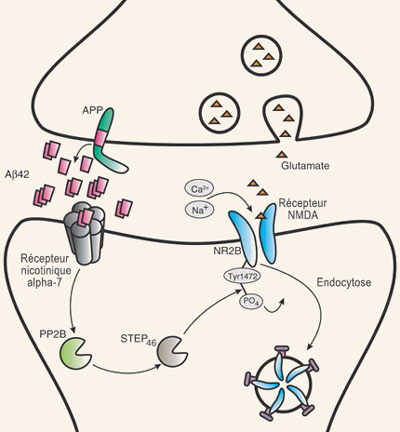
De nombreuses observations associent l’agrégation en plaques de fibrilles de bêta-amyloïde aux déficits cognitifs de l’Alzheimer. Cependant, le déploiement spatio-temporel de ces plaques amyloïdes dans le cerveau vieillissant est plutôt mal corrélé avec l’apparition progressive des pertes de mémoire et autres symptômes de l’Alzheimer. Sans parler des individus cognitivement normaux qui développent des plaques amyloïdes sans aucune trace de dommages neuronaux associés à ces plaques. Voilà pourquoi la recherche sur les mécanismes moléculaires de l’Alzheimer tend à délaisser les dépôts extracellulaires insolubles des plaques amyloïdes pour s’intéresser à la forme oligomérique soluble de la bêta-amyloïde. Et en particulier au rôle pathologique que cette forme non fibrillaire de la bêta-amyloïde pourrait jouer au niveau synaptique lorsqu’elle est produite en trop grandes quantités (car elle aurait aussi un rôle physiologique encore mal connu à des concentrations moindres). Ce modèle explicatif de l’Alzheimer où l’on considère comme centrale la toxicité synaptique des oligomères bêta-amyloïdes plutôt que celle des fibrilles agglutinées dans les plaques a reçu le nom d’hypothèse synaptique bêta-amyloïde. Celle-ci s’appuie sur au moins deux phénomènes bien avérés : la forte corrélation entre le degré de sévérité clinique de l’Alzheimer et la perte de synapses; et le taux de bêta-amyloïde soluble qui s’élève dans le cortex avec la progression des signes pathologiques chez les modèles animaux. Avec ces modèles animaux de l’Alzheimer, on a pu d’ailleurs faire plusieurs observations indiquant que les synapses pourraient être affectées négativement par cette hausse des oligomères de bêta-amyloïde. Des concentrations physiologiques de dimères et de trimères de bêta-amyloïde (mais pas de monomères) induisent par exemple une perte progressive de synapses dans l’hippocampe. D’autres études, chez la souris, ont montré que des pertes synaptiques réduisant l’efficacité de la PLT peuvent être observées avant même l’apparition de plaques amyloïdes. Des oligomères de bêta-amyloïde de faible poids moléculaire et de nature non fibrillaire peuvent aussi bloquer la potentialisation à long terme (ou PLT), l’un des mécanismes moléculaires à la base de l’apprentissage et de la mémoire. Ces oligomères sont également nécessaires et suffisants pour perturber de façon transitoire des comportements appris.
L’exposition prolongée à des taux élevés d’oligomères de bêta-amyloïde entraîne aussi un rétrécissement des épines dendritiques, ces bourgeonnements sur les dendrites des neurones qui forment la partie post-synaptique de la synapse. Cette baisse de densité des épines dendritiques est accompagnée par une diminution du niveau de débrine, une protéine du cytosquelette qui module la plasticité synaptique, avec le filament d’actine. Détail intéressant, cette diminution peut être bloquée par la mémantine, un médicament commercialisé sous le nom de Namenda et prescrit aux patients souffrant d’Alzheimer. Tout comme l’administration d’anticorps à la bêta-amyloïde empêche également cette détérioration des épines dendritiques.
Vers la fin des années 2000, des travaux ont également établi un lien possible entre la mort neuronale typique de l’Alzheimer et un mécanisme d’élimination des connexions neuronales excédentaires qui prédomine au tout début du développement cérébral. L’hypothèse ici est que ce mécanisme pourrait être réactivé par des processus liés au vieillissement. Ceux-ci impliqueraient non pas la bêta-amyloïde elle-même, mais plutôt la libération du fragment de l’APP adjacent à la bêta-amyloïde. Ce fragment, dit N-terminal, déclencherait la cascade de réactions moléculaires délétères en se fixant sur un récepteur appelé DR6 (pour « cell death receptor 6 », en anglais). Or ce récepteur DR6, fortement exprimé dans les régions cérébrales affectées par l’Alzheimer, est connu pour mettre en marche le phénomène de mort cellulaire programmée, ou apoptose (voir capsule outil à gauche). De plus, en bloquant l’activité du récepteur DR6, on a pu montrer que la dégénérescence axonale était retardée in vitro, et que les synapses redondantes demeuraient en place dans certaines régions du cerveau de souris. D’où l’hypothèse que l’activation du récepteur DR6 par le fragment N-terminal de l’APP réactiverait des mécanismes de mort cellulaire programmée normalement actifs au tout début du développement cérébral. Dans ce modèle, la bêta-amyloïde jouerait un rôle complémentaire en dégradant plutôt les synapses.
Un autre modèle proposé à la même époque que le précédent fait remonter la cause première de l’Alzheimer au-delà des plaques amyloïdes, à un dérèglement du processus de division cellulaire. Il s’agit donc ici aussi d’une réactivation tardive d’un processus ayant cours normalement très tôt durant le développement, celui de la différentiation des cellules souches en neurones. Mais les neurones matures et bien différenciés au niveau de leurs dendrites et de leur axone ne sont évidemment plus adaptés à la division cellulaire. Par conséquent, la réactivation des processus à l’origine de celle-ci serait fatale aux neurones du cerveau des adultes souffrant d’Alzheimer. |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

|
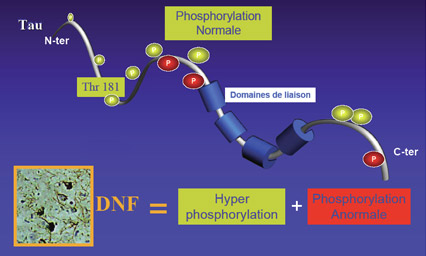
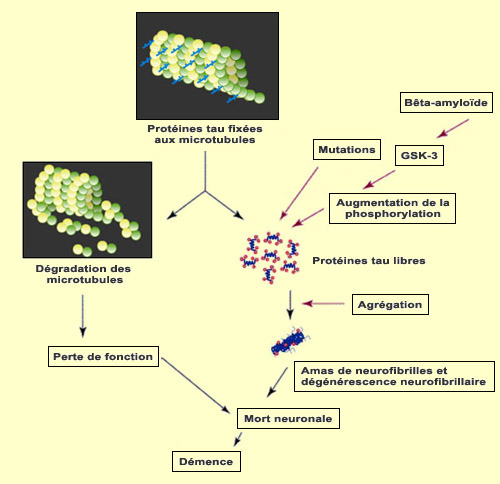
L’hypothèse de la cascade amyloïde a dominé la recherche sur l’Alzheimer pendant plus d’une décennie. Selon cette hypothèse, l’Alzheimer serait causée par l’accumulation de plaques amyloïdes (ou d’oligomères de bêta-amyloïdes) qui provoquerait les dégénérescences neurofibrillaires puis la mort neuronale. Mais depuis le début des années 2000, plusieurs données sont venues fragiliser cette hypothèse au profit d’explications alternatives. La plus connue est sans doute celle proposant plutôt comme élément premier dans la pathologie l’agrégation des protéines tau produisant les dégénérescences neurofibrillaires. Car sans les protéines tau pour les stabiliser, les microtubules se désagrègent, ce qui perturbe le transport axonal et finit par tuer le neurone. Et la perte neuronale est très fortement corrélée à la gravité des déficits cognitifs chez les patients Alzheimer. En ce qui concerne les mécanismes responsables de ce détachement des protéines tau des microtubules, plusieurs pensent que l’augmentation de la phosphorylation de cette protéine joue un rôle important. On a en effet observé que les protéines tau agglutinées sont très phosphorylées et que la phosphorylation des protéines tau réduit la force du lien qui les unit aux microtubules. La phosphorylation est
toutefois un phénomène complexe impliqué dans la régulation
de plusieurs processus, notamment du développement neuronal. Si bien que
le rôle précis de ce phénomène dans les
tauopathies comme l’Alzheimer est encore discuté. Certaines données
portent par exemple à penser que la phosphorylation des protéines
tau survient après l’agrégation et que ce sont davantage des
changements structuraux de la protéine qui sont associés à
son détachement et à son agrégation. On doit ici mentionner que ce ne sont pas tous les sites de phosphorylation identifiés sur la protéine tau qui sont impliqués dans la régulation de la liaison aux microtubules. Parmi ceux ayant une influence sur la liaison aux microtubules, il y a par exemple le site Thr181 (pour le 181e acide aminé qui est une thréonine) et les Ser202, 214, 262, 324 et 356 (pour l’acide aminé sérine à ces positions respectives).
L’état de phosphorylation des protéines tau dépend de la balance entre l’activité de deux types d’enzyme : les protéines kinases (qui ajoutent un groupement phosphate), et les phosphatases (qui enlèvent un groupement phosphate). D’un côté, on aura donc des kinases comme la protéine kinase A, la phosphorylase kinase ou la glycogène-synthétase-kinase 3. Et de l’autre les sérines et les thréonines phosphatases dont les activités antagonistes vont aussi contribuer à réguler l’activité de la protéine tau. L’étude de ces interactions enzymatiques sur le niveau de la phosphorylation des protéines tau in vivo est cependant très difficile pour plusieurs raisons. D'abord, l’activité des kinases in situ est souvent dépendante de leur propre état de phosphorylation. Autrement dit, l’activité des kinases susceptibles de phosphoryler la protéine tau est elle-même dépendante de l’activité d’autres kinases. Ces dernières, qui font partie d’autres cascades de réactions biochimiques peuvent donc influencer indirectement l’état de phosphorylation des protéines tau bien qu’elles n’interagissent pas directement avec elles. D’autre part, les phosphatases peuvent aussi déphosphoryler et donc inactiver des protéines kinases susceptibles de phosphoryler les protéines tau. On a donc encore ici des actions indirectes possibles. Plusieurs études montrent par ailleurs que des cofacteurs modulent également l’état de la protéine tau en augmentant ou réduisant sa phosphorylation. On peut mentionner ici les niveaux intracellulaires de calcium, d’AMP cyclique ou de phospholipides qui influencent des kinases comme la protéine kinase A. Ces cofacteurs modifient probablement la structure tridimensionnelle des protéines tau, les rendant possiblement de meilleurs substrats pour certaines kinases. Comme si cela n’était pas déjà assez complexe, on sait que la phosphorylation des protéines tau peut également être modulée par l’état physiologique de la cellule. Ainsi, dans des conditions de stress cellulaire, la phosphorylation des protéines tau est nettement accrue.
Cela étant dit, des candidats enzymatiques précis commencent tout de même à retenir l’attention. C’est souvent le cas d’enzymes situées en amont de la cascade biochimique qui favoriseraient à la fois l’agrégation des protéines tau et les plaques amyloïdes. C’est le cas de la glycogène-synthétase-kinase 3 (ou GSK-3), une protéine kinase responsable de la phosphorylation de plusieurs protéines dont la protéine tau. L’intérêt pour la GSK-3 vient du fait qu’en plus de son action sur la protéine tau, elle régule aussi le clivage de l’APP (le précurseur de la bêta-amyloïde), réduit la neurogenèse et augmente l’apoptose (voir capsule outil à gauche). Des phénomènes associés de près aux déficits cognitifs de l’Alzheimer. On a également découvert des mutations sur le gène de la protéine tau. Celles-ci donnent lieu à des protéines tau qui s’agglutinent davantage et se lient moins bien aux microtubules. Il s’agit donc encore d’un phénomène favorisant le détachement des protéines tau et leur agrégation qui produit la dégénérescence neurofibrillaire. |
| |
|
|
|
|
|
|
|
|