|
|
| Un livre accompagne le site depuis octobre 2024. Découvrez-le ici > | |
|
|
|
|
|
|||||
|
|
|||||||
|
|
|
|

| |
Des oligomères pour maintenir la trace de nos souvenirs
| |
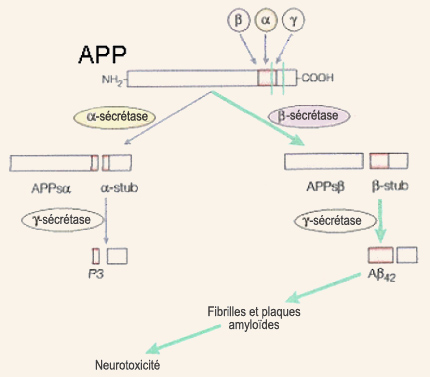
Déjà décrites par Alois Alzheimer au début du XXe siècle, les plaques amyloïdes n’ont commencé à livrer leurs secrets qu’en 1984 avec la caractérisation du peptide bêta-amyloïde, leur constituant principal, par George Glenner et son équipe. La bêta-amyloïde, et son précurseur l’APP (« Amyloid Protein Precursor », précurseur de la protéine amyloïde, en anglais), sont bientôt devenus au cœur de l’hypothèse de la cascade amyloïde formulée par Hardy et Higgins en 1992. Il s’agit de l’hypothèse la plus célèbre sur l’origine de la démence de type Alzheimer. Elle considère la production puis l’agrégation de la bêta-amyloïde comme le phénomène à l’origine des dérèglements dont souffrent les personnes atteintes d’Alzheimer. Les pathologies associées à la protéine tau, ainsi que d’autres hypothèses explicatives, sont alors considérées comme étant des processus en aval de cette cascade amyloïde. Le gène de la protéine précurseur de la bêta-amyloïde (ou APP) est situé sur le chromosome 21, et s’exprime à peu près dans tous les tissus de l’organisme. Ce gène, après copie en ARN messagers agencés de différentes façons, conduit à la production de glycoprotéines dont la longueur peut varier de 695 à 770 acides aminés, avec la forme à 695 acides aminés qui prédomine dans les neurones du cerveau. L’APP fait partie de la grande famille des protéines dites transmembranaires, car elle traverse la membrane cellulaire du neurone : sa longue partie N-terminale est située dans la partie extracellulaire et sa courte région C-terminale dans le cytoplasme. La protéine précurseur de la bêta-amyloïde (APP) peut donc, comme son nom l’indique, être coupée par des enzymes pour donner d’autres protéines plus petites, dont la bêta-amyloïde.
On parle d’une voie métabolique « amyloïdogénique » pour cette dernière et d’une voie « non-amyloïdogénique » pour la première qui serait donc bénéfique puisqu’elle empêcherait l’APP de former de la bêta-amyloïde. Selon l’hypothèse de la cascade amyloïde, un dysfonctionnement dans la voie amyloïdogénique entraînerait la production accrue de la forme longue du peptide bêta-amyloïde, celle à 42 acides aminés. On sait en effet que cette forme à 42 acides aminés, contrairement à la forme normale qui n’en compte que 40, s’agglutine plus facilement en plaques amyloïdes. Celles-ci contiennent d’ailleurs surtout la forme à 42 acides aminés durant les stades légers et modérés de l’Alzheimer. Ce n’est que tardivement dans l’évolution de cette démence que la forme à 40 acides aminés va également se retrouver dans les plaques. C’est donc l’agrégation de bêta-amyloïde à 42 acides aminés sous forme de fibrilles (grâce à la juxtaposition de leurs feuillets bêta), puis des fibrilles en plaques, qui serait toxique pour les neurones en permettant une trop grande entrée de calcium dans ceux-ci, entraînant leur mort par nécrose ou apoptose. La réaction inflammatoire concomitante, qui se traduit par la sécrétion de radicaux libres neurotoxiques par les cellules du système immunitaire, accentuerait cet effet létal. Les plaques de bêta-amyloïde induiraient donc toute la pathologie subséquente associée à l’Alzheimer, incluant la dégénérescence neurofibrillaire. Cette hypothèse amyloïde, longtemps le paradigme dominant de la recherche sur l’Alzheimer, a montré ses limites et est aujourd’hui débattue. Un certain nombre de faits avérés continuent toutefois de plaider en sa faveur. C’est le cas de la découverte par l’équipe de John Hardy, en 1991, que certains cas de la forme familiale de l’Alzheimer sont provoqués par des mutations sur l’APP. De plus, ces mutations conduisent à une production accrue de la forme longue de la bêta-amyloïde, plus propice aux agrégats. Voilà donc deux observations appuyant l’idée que l’origine de la cascade d’événements menant à l’Alzheimer remonte à des mutations affectant la production de bêta-amyloïde. Les cas les plus fréquents de la forme familiale de l’Alzheimer proviennent toutefois de mutations sur le gène préséniline 1 (PS1), situé sur le chromosome 14, ainsi que sur un gène analogue, nommé PS2, situé sur le chromosome 1. En 1996, différentes équipes ont montré que ces les protéines PS1 et PS2 mutées favorisent elles aussi la forme longue de la bêta-amyloïde, probablement en interagissant avec le catabolisme de l’APP. Tout porte à croire également que les protéines transmembranaires issues des gènes de la préséniline 1 et 2 seraient derrière l’activité enzymatique des gamma-sécrétases, dont le site de coupure sur l’APP est justement quelque part au mileu de la membrane cellulaire. Sans être déterminé par des mutations autosomales dominantes comme pour la forme familiale, l’Alzheimer dite sporadique n’en est pas moins influencée par certains gènes dont celui de l’apolipoprotéine E (ou ApoE) est le plus clairement impliqué (voir l’encadré). En 1993, l’équipe de Judes Poirier a en effet démontré que la présence de l’une des trois formes principales de la protéine ApoE, l’ApoE4, constitue un facteur de risque tant pour la forme familiale que sporadique de l’Alzheimer. Différents mécanismes sont possibles pour expliquer cet effet néfaste de la variante ApoE4, dont une diminution de l’élimination du peptide bêta-amyloïde. L’ApoE semble aussi être impliqué dans la formation des fibrilles amyloïdes qui s’agglutinent en plaques. Il est d’ailleurs considéré comme un cofacteur de l’amyloïdogenèse. D’autres études chez la souris ont démontré que l’ApoE4 diminuait la complexité de l’arbre dendritique des neurones corticaux ainsi que la densité des épines dendritiques. D’autres suggèrent encore que la protéine ApoE4 est moins performante dans la réparation neuronale et qu’elle perturbe l’induction de la potentialisation à long terme dans l’hippocampe. Un autre facteur génétique qui pourrait prédisposer à l’Alzheimer et qui attire de plus en plus l’attention est le gène de la clusterine, aussi connue sous le nom de Apolipoprotéine J. Il s’agit d’une protéine versatile qui possède plusieurs propriétés de protection et de réparation d’autres protéines mal repliées. Elle peut ainsi se lier au peptide bêta-amyloïde et prévenir la formation des fibrilles à l’origine des plaques. Elle est également impliquée dans l’élimination des peptides bêta-amyloïdes et de leurs fibrilles. On sait depuis la fin des années 1980 que l’expression de la clusterine est augmentée chez les patients Alzheimer, comme si l’organisme essayait par là de prévenir la progression des plaques amyloïdes. Vers la fin des années 2000, deux consortiums internationaux ont également comparé près de 600 000 marqueurs génétiques chez plus de 20 000 personnes et ont confirmé l’importance que semble jouer la clusterine dans l’Alzheimer. Outre ces données mettant en évidence la production et à l’agrégation de la bêta-amyloïde, il y a toutefois plusieurs observations qui sont difficiles à réconcilier avec l’hypothèse de la cascade amyloïde. Par exemple, même si les souris ayant les mutations de la forme familiale de l’Alzheimer produisent la forme à 42 acides aminés de la bêta-amyloïode en excès (et développent les plaques amyloïdes correspondantes), la plupart ne montrent pas de pertes neuronales significatives, ont peu de phosphorylation de la protéine tau, et pas de dégénérescences neurofibrillaires comme le prédit l’hypothèse amyloïde. Et la même absence de correspondance s’observe dans certaines régions du cerveau humain comme le cervelet qui peut compter beaucoup de plaques amyloïdes, mais pas les phénomènes associés selon l’hypothèse amyloïde. La controverse autour de cette hypothèse est aussi grandement alimentée par les mauvaises corrélations entre la dynamique spatiale et temporelle de la formation des plaques et la sévérité des déficits cognitifs observés. À l’opposé, le déclin cognitif dans l’Alzheimer est très bien corrélé avec la perte synaptique. Cela est particulièrement intéressant en regard des études concernant le rôle de la bêta-amyloïde sous sa forme non fibrillaire soluble dans la destruction des synapses.
|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
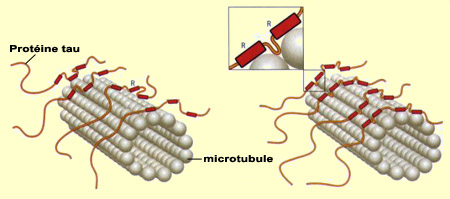
Les neurones ne peuvent survivre si les nutriments et autres éléments essentiels au maintien de leur structure ne peuvent circuler à l’intérieur de ceux-ci, en particulier dans leurs longs prolongements. Ce transport nécessite tout un réseau de microtubules qui, tels les rails d’un chemin de fer, prennent en charge les substances jusqu’à la bonne destination. Ces neurones en santé peuvent alors communiquer entre eux et assurer les fonctions cognitives propres à l’être humain. Or quand les « traverses » de ces rails de microtubules se défont, le réseau de transport interne des neurones défaille et entraîne bien souvent la mort du neurone avec lui. À grande échelle, cette mort neuronale entraîne la déstabilisation de tout le réseau de communication neuronal du cerveau, et des déficits cognitifs parfois graves, comme dans le cas de l’Alzheimer. Le concept de pathologie tau ou « tauopathies » désigne l’ensemble des phénomènes qui résultent du dysfonctionnement des protéines tau, les fameuses « traverses » qui stabilisent normalement les microtubules. Les six variantes de la protéine tau sont synthétisées à partir du même gène, situé sur le chromosome 17. Ce gène peut cependant être lu de six manières différentes, d’où autant d’ARN messager et de variantes de la protéine. La protéine tau se distingue notamment par la répétition d'un motif particulier dans sa séquence d’acides aminés, noté R (pour « Repeat », en anglais). C’est cette région R qui permet à la protéine tau de s’arrimer aux microtubules. Trois des variantes tau possèdent ce motif trois fois, et les trois autres le répètent quatre fois. Par conséquent, on parle de variantes 3R ou 4R. Ayant un point d’ancrage de plus, les protéines tau 4R stabilisent mieux les filaments de microtubules à l'intérieur du neurone que les variantes 3R, favorisant des prolongements plus longs et plus rigides. La forme des neurones peut ainsi être façonnée en fonction du type de variante des protéines tau exprimé.
En plus du type de variante de la protéine tau exprimé (3R ou 4R), la stabilisation des microtubules peut être modulée encore plus finement par le niveau de phosphorylation de la protéine tau. La phosphorylation est un processus par lequel un groupement phosphate se lie à certains acides aminés d’une protéine. Or la protéine tau comporte de nombreux sites de phosphorylation et l’on observe que plus il y a de groupements phosphates qui s’y fixent, moins la protéine tau interagit avec les microtubules, jusqu’à s’en détacher complètement. Les protéines tau ainsi inactivées par l'excès de phosphorylation s'associent alors entre elles pour former des paires de filaments hélicoïdaux. Ceux-ci s’associent à leur tour pour produire les dégénérescences neurofibrillaires qui envahissent progressivement le neurone et perturbent son fonctionnement jusqu’à le détruire. Quant à savoir ce
qui serait à l’origine de l’augmentation de la phosphorylation
de la protéine tau, cela demeure une question ouverte. Une hypothèse
soutien par exemple que ce sont les radicaux
libres, dus à la présence de la bêta-amyloïde,
qui seraient les coupables en détériorant la membrane cellulaire
du neurone. Des ions calcium et des fragments de bêta-amyloïde pourraient
alors pénétrer plus facilement dans le neurone et suractiveraient
certaines protéines kinases, dont certaines contrôlent la phosphorylation
des protéines tau. |
| |
|
|
|
|
|
|
|
|